Software

MVNpermute: An R package for doing valid permutation-based testing when samples are correlated (as happens when there are related subjects). The package generates a new set of “permuted” outcome variables (e.g. traits) from which a properly calibrated p-value can be estimated. Available on CRAN.
Multipoint IBD estimation with LD: A hidden Markov based model that can compute multipoint estimates of IBD given dense genotype data (i.e. in the presence of linkage disequilibrium). Given dense genotype data we can recover essentially the true amount of chromosome-wide IBD sharing between a pair of individuals and get very accurate IBD estimates at arbitrary genetic positions. Our method is fast (it can estimate IBD genomewide for a single pair given ~300,000 SNPs in under ~30 seconds) and is robust to pedigree errors. The current software can use a pedigree of essentially arbitrary size and complexity, if available, or base the estimates given only genomic data (this latter method is called GIBDLD).
The current version of IBDLD is 3.x and is available here. IBDLD 3.x has been further optimized for speed, including the ability to run in parallel. Additional model refinements have also significantly increased accuracy.
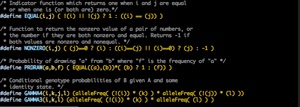
Identity Coefficients: Compute the condensed identity coefficients for pairs of individuals (Kan Lange’s book Mathematical and Statistical Methods for Genetic Analysis [2002] has a good description of identity coefficients). This software uses novel algorithms I developed to be able to do the computations very efficiently and rapidly, even for very large and complex pedigrees.
The current version is IdCoefs 2.1.
IBD Estimation and Linkage: Estimate pairwise identity by descent (IBD) probabilities in very large pedigrees. This software can compute the proportion of alleles shared IBD between a pair of individuals at a marker given all genotypes at that marker and the entire, unbroken, pedigree. The method implemented here is particularly useful for pedigrees that are very deep and there are multiple generations with missing genotype data. This requires knowing the condensed identity coefficients of a subset of the individuals in the pedigree.
The current software version is RAPID 1.0.2.
Genetic Analysis of Qualitative Traits: Estimate covariate effects and variance components for a binary trait. Here we use a generalized linear mixed model for the trait and use a Monte Carlo expectation maximization method to obtain maximum likelihood estimates of the model parameters. Our method can successfully analyze large samples (hundreds) of affected and unaffected individuals, all of whom may be related.
The current version of BVC is available here.
Genomewide Association of Binary Traits in Related Populations: Mixed model-based approaches to genome-wide association studies (GWAS) of binary traits in related individuals can account for non-genetic risk factors in an integrated manner. However, they are technically challenging. GLOGS (Genome-wide LOGistic mixed model/Score test) addresses such challenges with efficient statistical procedures and a parallel implementation. GLOGS has high power relative to alternative approaches as risk covariate effects increase, and can complete a GWAS in minutes.
The current version of GLOGS is here.
Updated February 4, 2016